Newly Diagnosed
Peroxisome Disorder Information and Resources
You have been told that you or your child has or may have a peroxisomal disorder. We understand this is a scary and difficult time for you and your family. We all have questions about the diagnosis and what it means for ourselves and loved ones. You may have some really difficult days ahead of you, but please remember, there are many of us who have been there and are here to support you. Please do not hesitate to reach out to The GFPD to be connected to others and receive guidance, resources, and support.
Here are some questions and responses from other families who have been impacted by peroxisomal disorders. Please keep in mind these are not a replacement for your doctor’s advice.
Peroxisomes are necessary for healthy cell function, normal brain development, and the formation of myelin. Peroxisomes are organelles inside every cell of the body that contain enzymes and help carry out cellular functions.
Peroxisomal disorders are rare, genetic, terminal conditions that affect all major organ systems of the body.

You may have been told that you or your child has Zellweger Syndrome (ZS), Neonatal Adrenoleukodystrophy (NALD), Infantile Refsum Disease (IRD), or Heimler syndrome.
As the understanding of peroxisomal disorders has grown, there has been a movement away from the use of how these were historically described.
It is now recommended to replace these names with the overall classification of this spectrum by using the terminology, Peroxisome Biogenesis Disorder-Zellweger Spectrum Disorder (PBD-ZSD).
You may also see us simply use peroxisomal disorders to be inclusive of all phenotypes the GFPD supports, including, D-Bifunctional Protein Deficiency (DBPD), Acyl-CoA Oxidase Deficiency (ACOX), and Alpha-Methylacyl-CoA Racemase (AMACR) Deficiency.
While there is currently no cure for peroxisomal disorders, treatment is symptomatic. It is recommended that individuals diagnosed with peroxisomal disorder take a multivitamin supplement which includes higher levels of vitamins A, D, E, and K. Some Individuals experience coagulopathy (impaired blood clotting) and therefore require additional supplementation of vitamin K (e.g., Mephyton).
Many individuals also may take a DHA supplement. Individuals with adrenal insufficiency will require daily steroid replacement treatment as prescribed by their endocrinologist. Individuals experiencing seizures will require anti-seizure medicine as prescribed by their neurologist. Individuals may also experience spontaneous bone fractures, as well as osteopenia. An orthopedic specialist, endocrinologist, or a combination of both may advise families on the best treatment for bone loss and/or fractures.
More information about treatments for the peroxisomal disorder, Zellweger Spectrum Disorder, can be located in the Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines.
Reminder: This information is not to take the place of your child’s primary care physician and medical team.
The Zellweger spectrum disorder: A cross-sectional study of symptom prevalence using input from family caregivers publication is the largest one to date on this population of patients and can provide a better idea of what symptoms are occurring in individuals affected with a peroxisomal biogenesis disorder, Zellweger spectrum disorder (PBD-ZSD).
In addition to a primary care physician/pediatrician, individuals with a peroxisomal disorder may need the following specialists: Audiologist, Endocrinologist, Gastroenterologist (GI), Geneticist, Hematologist, Neurologist, Ophthalmologist, Orthopedic, Otolaryngologist (ENT), Pulmonologist, and Urologist.
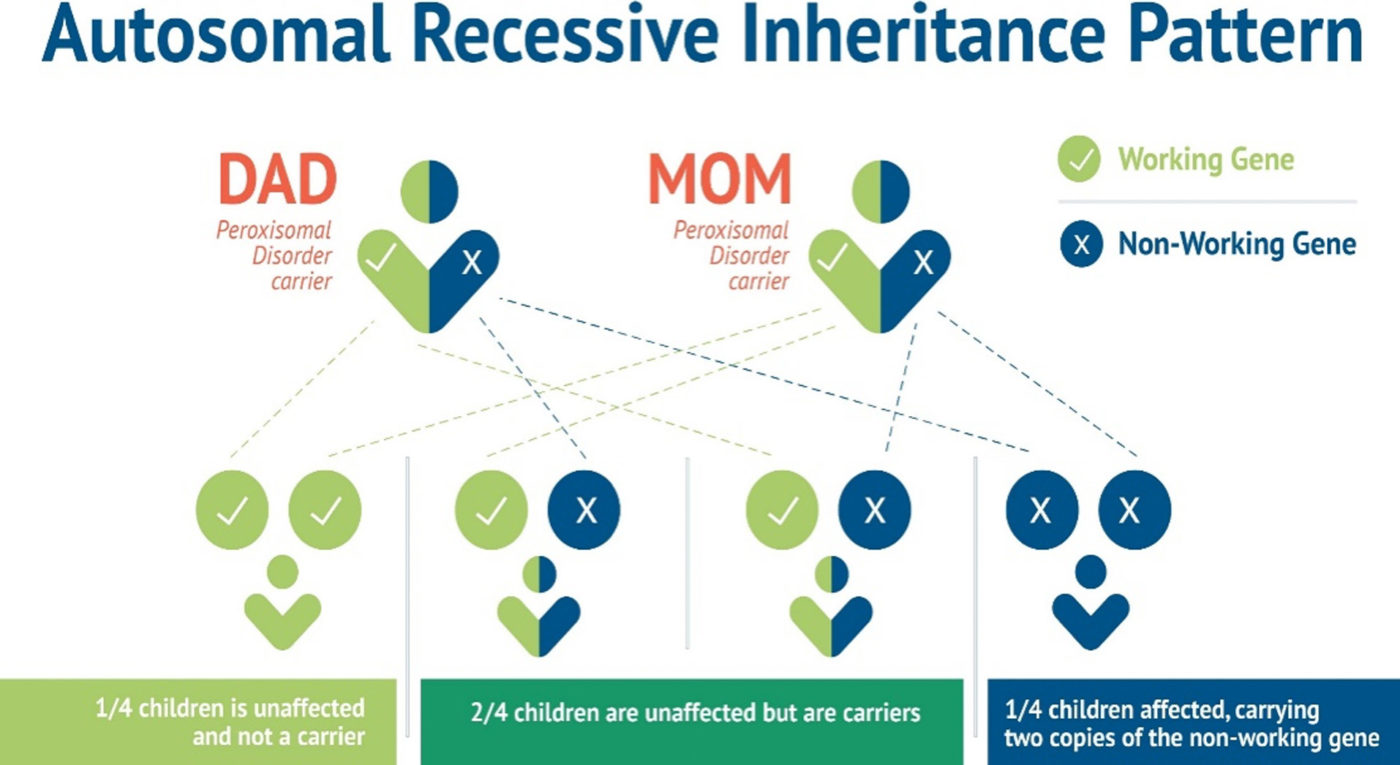
Peroxisomal disorders in the Zellweger spectrum and the related single enzyme protein deficiencies are inherited in an autosomal recessive pattern.
“To have an autosomal recessive disorder, you inherit two mutated genes, one from each parent. These disorders are usually passed on by two carriers.
Their health is rarely affected, but they have one mutated gene (recessive gene) and one normal gene (dominant gene) for the condition.
With each pregnancy, two carriers have a 25 percent chance of having an unaffected child with two normal genes (left), a 50 percent chance of having an unaffected child who is also a carrier (middle), and a 25 percent chance of having an affected child with two recessive genes (right).”
The Global Foundation for Peroxisomal Disorders, a 501(c)(3) public charity, currently connecting more than 600 families from over 40 countries with the following types of Peroxisomal Disorders: Zellweger Spectrum Disorder (ZSD), D-Bifunctional Protein Deficiency (DBPD), Acyl-CoA Oxidase Deficiency (ACOX), and Alpha-Methylacyl-CoA racemase (AMACR) Deficiency.
GFPD patients and families have an array of diverse experiences creating an invaluable source of support. The GFPD supports these families through connections to medical & scientific professionals, educational and scientific conferences, webinars, bereavement support, peer support, online support groups, advocacy, and provides an equipment exchange program. The GFPD shares objective and credible information to professionals, families, caregivers of patients, and individuals with peroxisomal disorders. The GFPD is a voice in the public arena for individuals affected by these rare disorders.